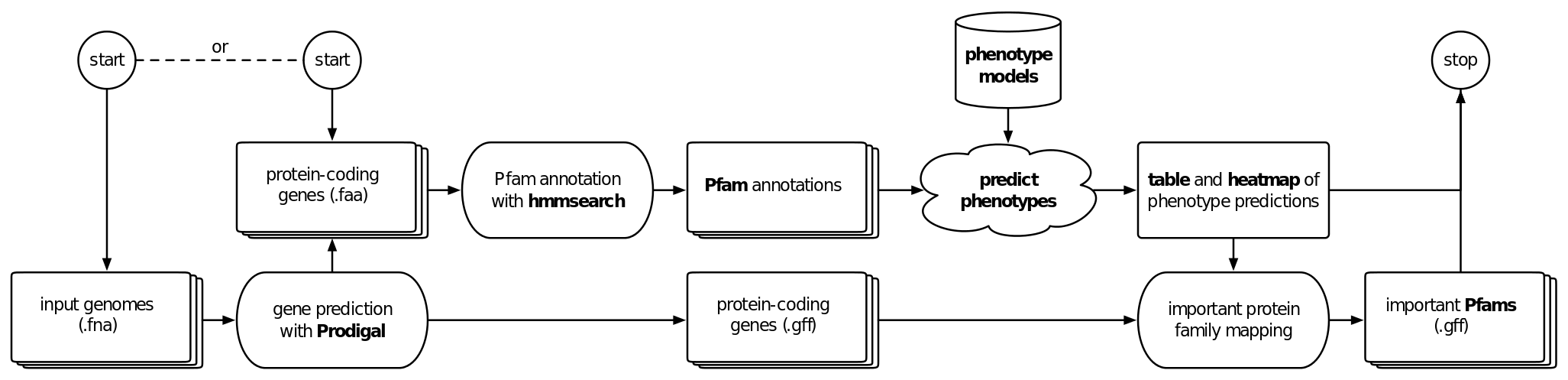
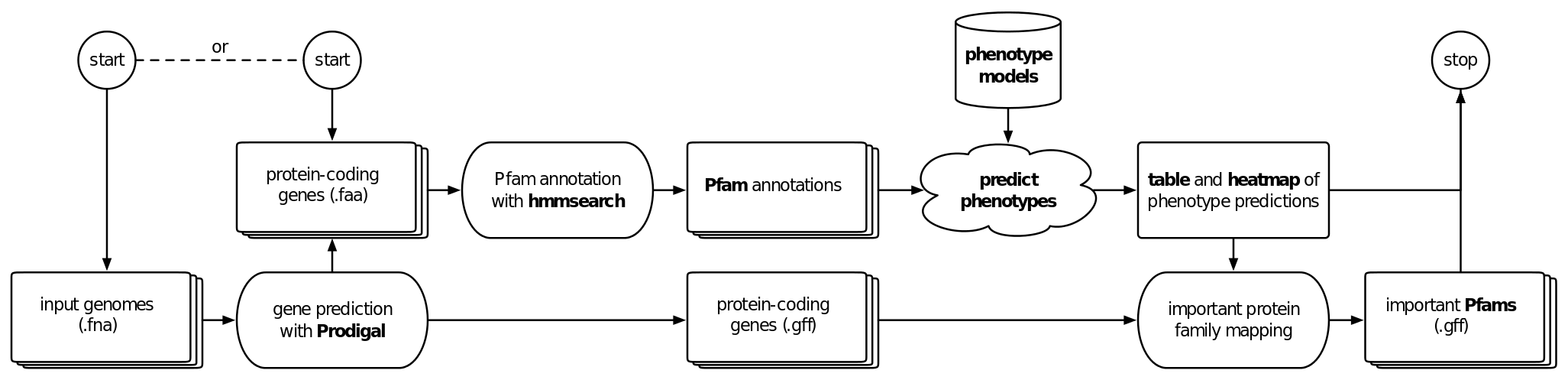
There is an increasing number of studies with a large number genomes recovered from isolate, metagenome, or single cell sequencing. To bridge the gap between the available genome sequences and available phenotype information, we have developed Traitar, a bioinformatics software to phenotype bacteria based on their genome sequence (see workflow below) . Traitar includes phenotype models for predicting 67 traits such as the use of different substrates as carbon and energy sources, oxygen requirement, morphology, and antibiotic susceptibility, and it provides the means to inspect the protein families (Pfams) that gave rise to these phenotype predictions.
In a paper recently published in mSystems (https://doi.org/10.1128/mSystems.00101-16), we describe the application of Traitar to two novel Clostridiales species with partical genomes recovered from metagenome shotgun sequencing of commercial biogas reactors. Traitar could verify an expert metabolic reconstruction and furthermore pinpoint additional traits that were missing in the manual metabolic reconstruction.
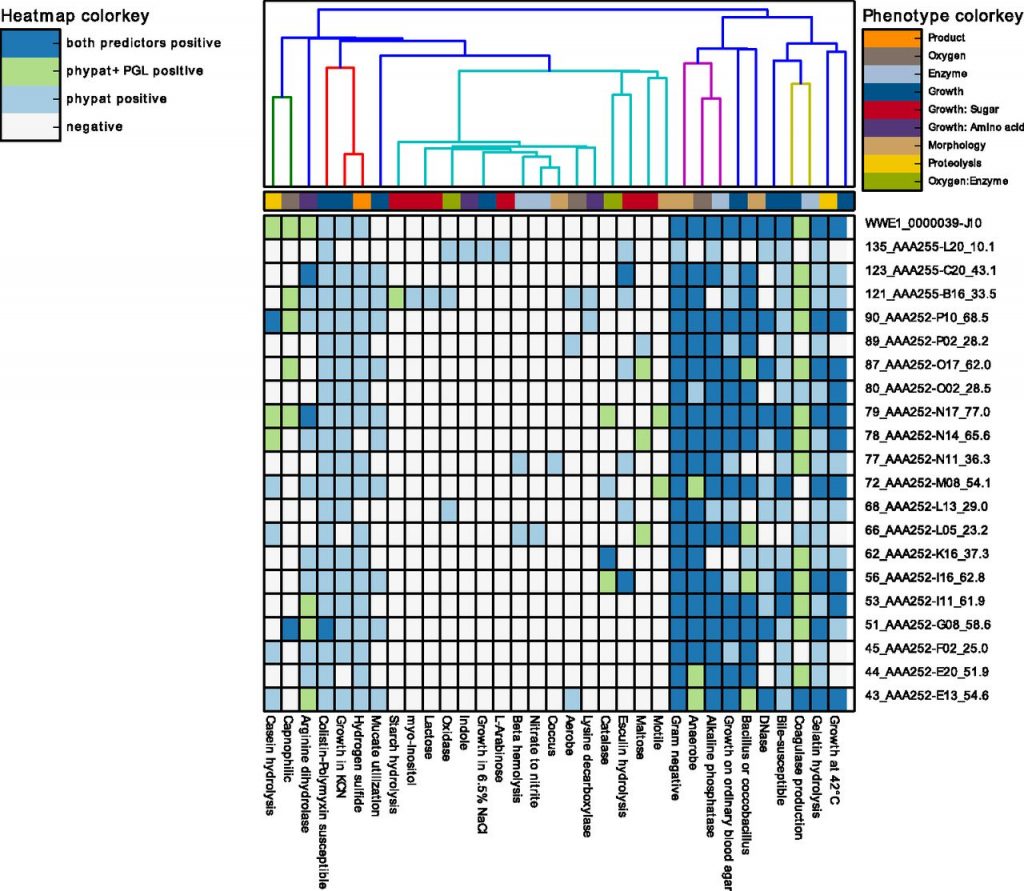
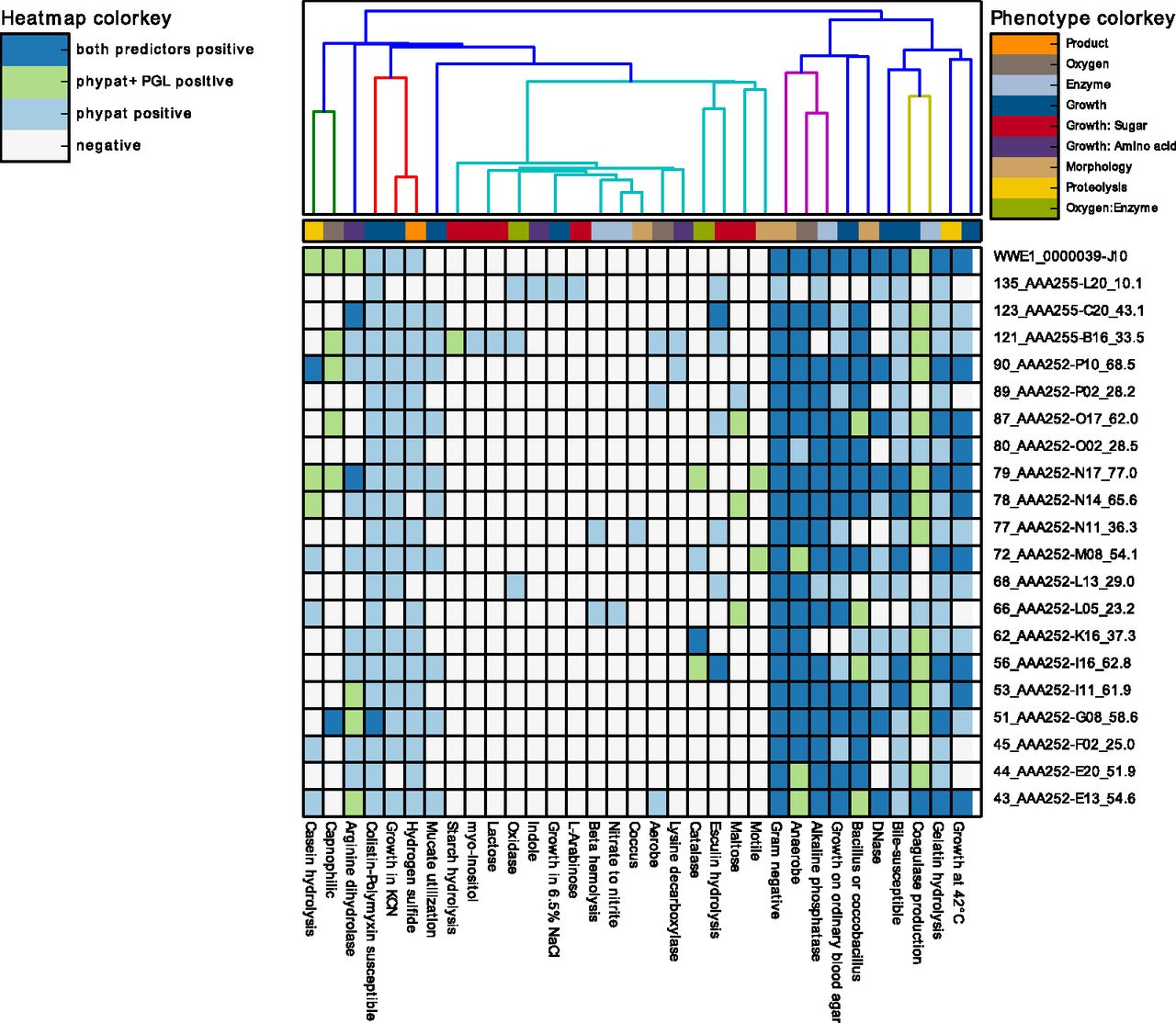
The software is easy to install and run. It only requires a nucleotide or protein FASTA file per sample as input. Users can inspect the phenotyping results of from Traitar for their genome sequences of two prediction modes (phypat and phypat+PGL) through heatmaps (see example of Traitar applied to single-assembled genomes below; Fig 5 in Traitar paper) and flat text files. For phenotyping a single genome, Traitar only requires a couple of minutes. Computation is multithreaded (parallelized) and scales to data sets with hundreds of genomes. We also offer a web service for data sets of up to around ten genomes. If you have larger data sets and troubles running the Traitar stand-alone tool get in touch with us (contact details below).
{kind=link}
To build and validate the phenotype models in Traitar, we have used phenotype data from the Global Infectious Disease and Epidemiology Online Network (GIDEON) and Bergey’s Systematic Bacteriology. Internally, the models were created using a machine learning method, namely L1 regularized L2 loss support vector machine trained on information about the presence and absence of protein families as well as ancestral protein family gains and losses.
Some word of advice when applying Traitar for phenotyping your genomes:
The training data from GIDEON and Bergey’s does not cover all known bacterial taxa and some with more data than others. Thus, some of the phenotypes might be realized with different protein families in taxa that are less well represented here and classification accuracy for these taxa be less than for others. Since Traitar provides the Pfam families responsible for your phenotype prediction, you could cross reference the phenotypes predicted by Traitar and the associated protein families with a targeted metabolic reconstruction approach.
We are currently working on incorporating new phenotypes and on further extending the existing phenotype models. For instance, we will apply Traitar to several hundred isolate genomes of the pathogen Pseudomonas aeruginosa to learn phenotype models of antibiotic resistance. We will keep updating the software and models, so please regularly check out our GitHub or Twitter. Traitar is designed to easily incorporate new prediction models. If you have data for phenotypes of interest please get in touch with us. We’re also preparing a stand-alone software to allow users to train their own phenotype models.
—
Aaron Weimann: @aaron_weimann
Andreas Bremges: @abremges
Alice C. McHardy: @alicecarolyn
GitHub: https://github.com/hzi-bifo/traitar
Web service: https://research.bifo.helmholtz-hzi.de/webapps/wa-webservice/pipe.php?pr=traitar
Web service and general BIFO software support: bifo-software@helmholtz-hzi.de