In scientific manuscripts, we tell stories of our research, generally in straight-line fashion with clear motivations and results. This type of research is rare (in my experience), with stories, motivations, and applications only realized post hoc. This is the nature of science, and our recent ISMEJ publication is no different.
With “16Stimator: statistical estimation of ribosomal gene copy numbers from draft genome assemblies“, we introduce an exciting method to generate 16S rRNA gene (16S) copy number estimates for bacterial genomes based on comparison of sequencing read depths of ribosomal and single copy gene regions. Application of this method resulted in 16S copy number estimates for hundreds of bacterial species without closed genome representatives. This extended database of known 16S copy numbers combined with phylogenetic based normalization methods (Kembel et al., 2012; Langille et al., 2013 – PICRUSt) for 16S amplicon sequencing studies will lead to more accurate organismal abundance measurements. (Note: article is not open access but the code is freely available)
These are valid and important motivations and applications, but really, we just wanted to know the 16S copy numbers for a handful of isolates so we could properly measure their abundances by amplicon sequencing in controlled community studies.
So here is the actual development route of 16Stimator:
A caveat of 16S amplicon sequencing studies is that, due to variation in bacterial 16S copy number, sequencing read and organismal abundances are not equivalent. For our controlled community experiments using leaf endophytic bacteria originally isolated from Arabidopsis thaliana, we needed to determine each isolate’s 16S copy number. We chose whole genome sequencing and assembly for this task. That was a horrible choice.
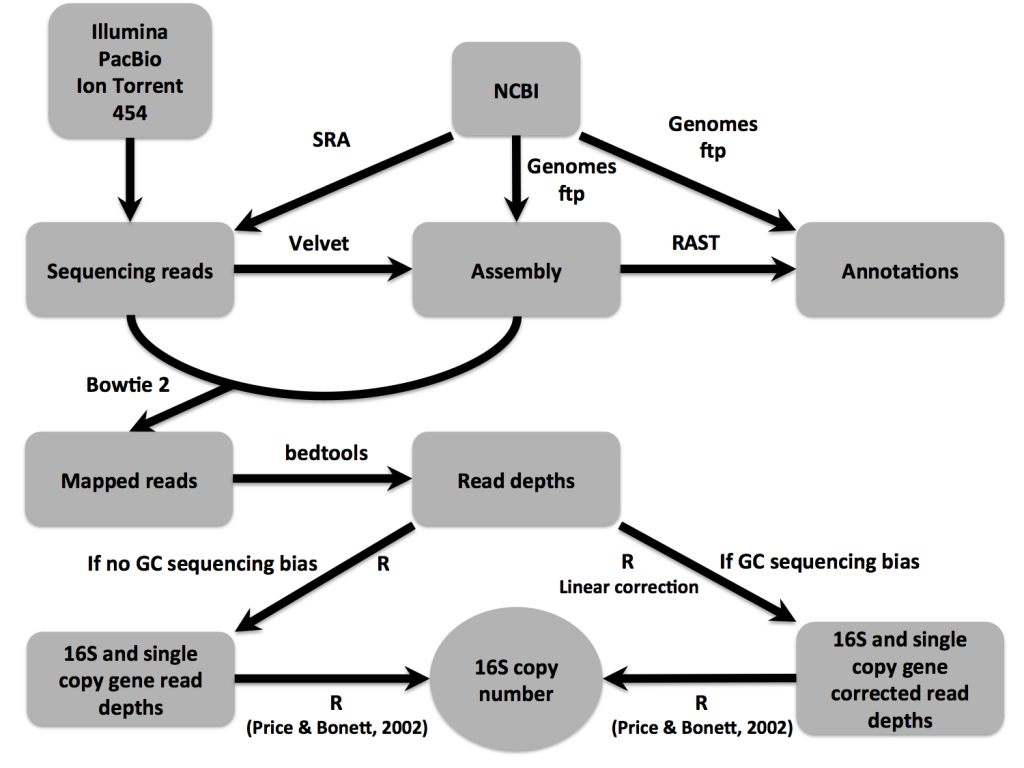
Current assembly algorithms do a poor job resolving repetitive genomic regions. Longer reads or larger insert sizes can overcome this limitation, but alas, we had short read, Illumina sequencing libraries with insert sizes smaller than ribosomal rRNA gene regions. After assembly, the 16S rRNA gene was found in one to few contigs. When we mapped reads back to the assembly, the coverage of the 16S contig was much greater than the average genomic coverage, so we sought to use read-depths to resolve 16S copy numbers. By statistical coverage comparisons of 16S to single copy, conserved genes, we were able to accurately estimate copy numbers.

Though the focus of the paper is on the sequencing read-depth approach, we did confirm 16S copy numbers experimentally, using an efficient qPCR approach. We compared amplification of 16S to single copy, conserved genes to determine copy number. The IDT-DNA gBlocks provided a convenient alternative to plasmid construction for creating standards with a 1:1 ratio of 16S to single copy gene.
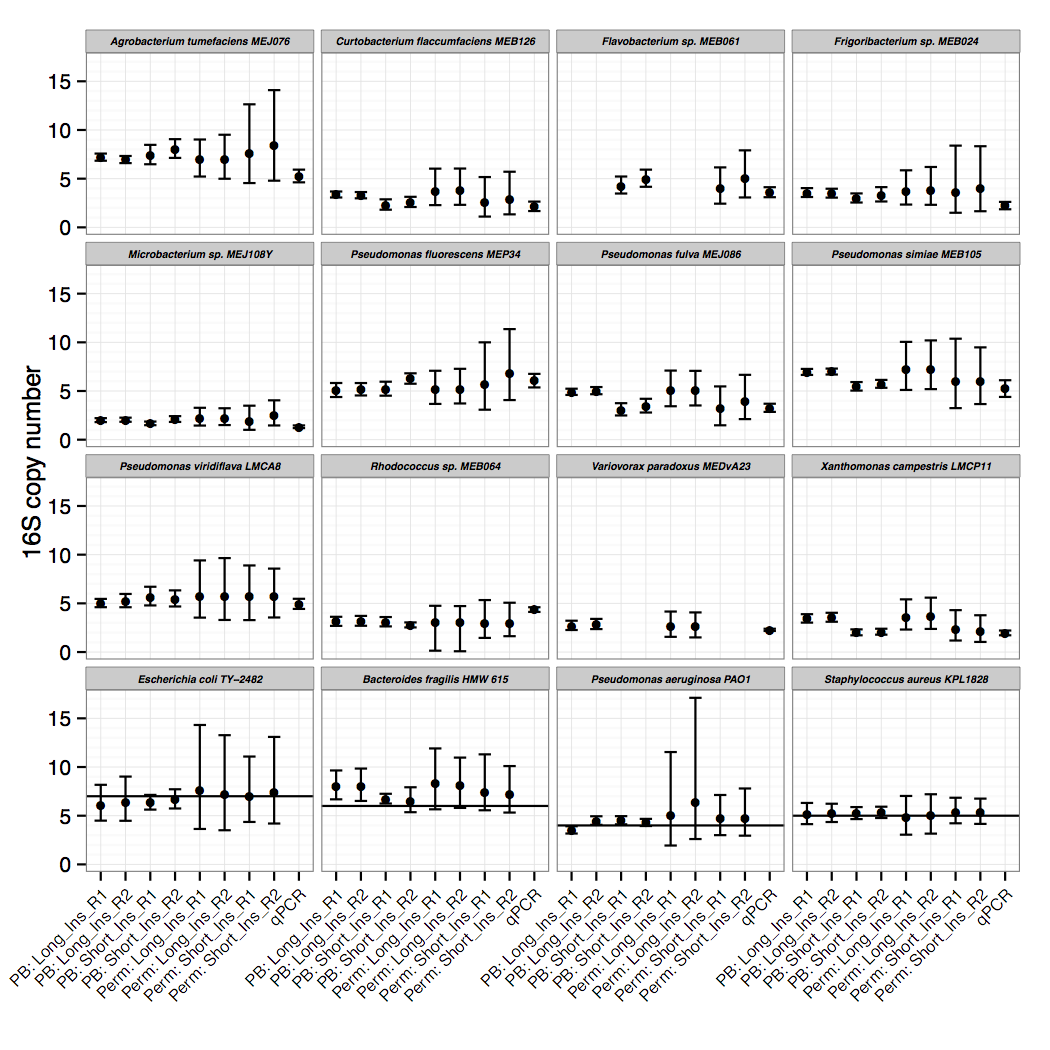
Only after resolving 16S copy numbers for our isolates of interest did we realize that this method could be applied to thousands of other draft genomes. We scaled 16Stimator to process tens of thousands of sequencing libraries deposited in SRA, resulting in 16S copy number estimations for hundreds of species without closed genome representatives. A large and diverse database of 16S copy numbers combined with methods to correct for copy number bias in 16S amplicon sequencing studies will ultimately result in more accurate abundance and diversity estimates. If sequencing reads are publicly deposited along with draft genome sequences, then the database can continue to grow.
Though we did not initially intend to create a method to estimate 16S copy numbers from draft genomes, science threw us a curveball and 16Stimator was our response. All the scripts and data are publicly available at https://bitbucket.org/perisin/16stimator. We look forward to feedback on our method to continue to improve and generate 16Stimates!
References:
- Kembel SW, Wu M, Eisen JA, Green JL. (2012). Incorporating 16S Gene Copy Number Information Improves Estimates of Microbial Diversity and Abundance. PLoS Comput Biol 8:e1002743.
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology 31:814—821.
- Perisin M, Vetter M, Gilbert JA, Bergelson J. (2015) 16Stimator: statistical estimation of ribosomal gene copy numbers from draft genome assemblies. ISMEJ. doi: 10.1038/ismej.2015.161
- Price RM, Bonett DG. (2002). Distribution-Free Confidence Intervals for Difference and Ratio of Medians. Journal of Statistical Computation and Simulation 72:119—124.