This seems like it could be of interest to microbial community researchers: The ISME Journal – High-resolution phylogenetic microbial community profiling
Abstract (with bolding by me)
Over the past decade, high-throughput short-read 16S rRNA gene amplicon sequencing has eclipsed clone-dependent long-read Sanger sequencing for microbial community profiling. The transition to new technologies has provided more quantitative information at the expense of taxonomic resolution with implications for inferring metabolic traits in various ecosystems. We applied single-molecule real-time sequencing for microbial community profiling, generating full-length 16S rRNA gene sequences at high throughput, which we propose to name PhyloTags. We benchmarked and validated this approach using a defined microbial community. When further applied to samples from the water column of meromictic Sakinaw Lake, we show that while community structures at the phylum level are comparable between PhyloTags and Illumina V4 16S rRNA gene sequences (iTags), variance increases with community complexity at greater water depths. PhyloTags moreover allowed less ambiguous classification. Last, a platform-independent comparison of PhyloTags and in silico generated partial 16S rRNA gene sequences demonstrated significant differences in community structure and phylogenetic resolution across multiple taxonomic levels, including a severe underestimation in the abundance of specific microbial genera involved in nitrogen and methane cycling across the Lake’s water column. Thus, PhyloTags provide a reliable adjunct or alternative to cost-effective iTags, enabling more accurate phylogenetic resolution of microbial communities and predictions on their metabolic potential.
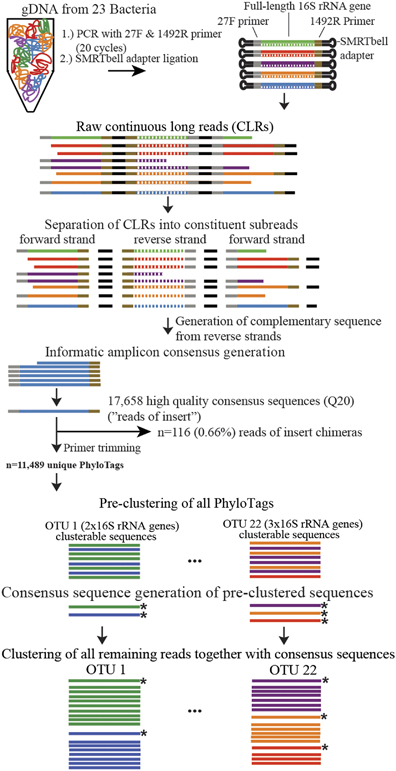
Basically, they use PacBio sequencing and a novel informatics workflow to generate full length 16S rRNA sequences from communities of microbes.
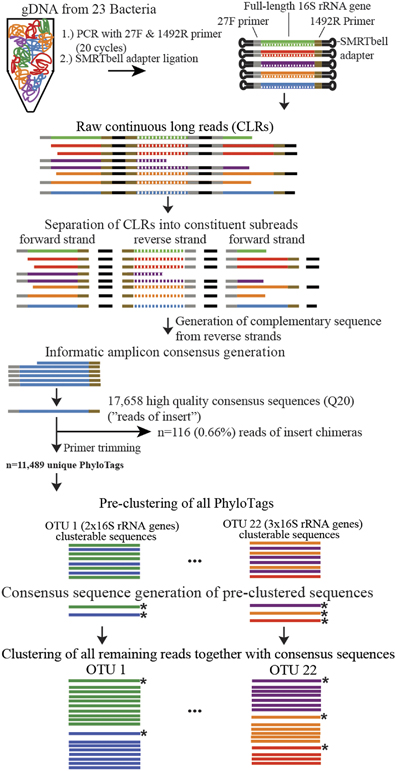
And they show this approach can be a powerful tool in characterizing communities, and perhaps most importantly allows one to more accurately phylogenetically analyze members of the community than short “iTag” sequencing. Now, this approach is more expensive and not everyone has access to a PacBio system, but it could be of use to others out there.
They conclude
Using PhyloTags to assess microbial community diversity in environmental samples allows us to fill important gaps in the tree of life while improving classification and microbial community profiling accuracy with important implications for inferred metabolic potential and biogeochemical roles of uncultivated microorganisms in natural and human engineered ecosystems
One issue that hopefully will get addressed:
The paper reports “Raw and processed sequence data is publically available on the JGI Genome portal page (http://genome.jgi.doe.gov/PhyloTag.html).” but that link is dead.